BEIJING, April 3, 2026 /PRNewswire/ -- Shanghai Fashion Week has risen to the fourth place globally, overtaking New York, according to a report released Thursday in Shanghai. The Global Fashion Industry Index-Fashion Week Vitality Index Report (2025), issued by China Economic Information Service, describes Shanghai as the fastest-growing node city in the global fashion landscape.
Paris and Milan fashion weeks retained the top two positions globally. London and Shanghai ranked third and fourth, with commercial vibrancy and digital transformation cited as their core competitiveness.
For the first time, the report added a "commercial trade activity" indicator to quantify each fashion week's ability to integrate commercial resources and convert them into market transactions. Shanghai Fashion Week recorded 1,217 trade brands, second only to Paris, and operated seven digital trade platforms which is the highest figure among all eight fashion weeks surveyed.
The report noted persistent gaps. Shanghai Fashion Week's 23 product categories and average transaction price of 420 U.S. dollars represent only 64 percent and 54 percent of Paris Fashion Week's figures respectively, reflecting a market still dominated by mid-range and emerging brands but with significant growth potential.
In the elements aggregation dimension, Paris and Milan led with 429 and 387 participating brands and 544 and 529 events respectively, while Shanghai hosted around 200 events and distinguished itself through new brand participation. In industry influence, Shanghai ranked first globally in the number of designers, and its innovation trend score also placed near the top.
China's apparel and fashion retail market reached nearly 460 billion U.S. dollars in 2025, maintaining its position as the world's largest single market. Global fashion e-commerce penetration rate rose from 18 percent in 2020 to approximately 36 percent in 2025, surpassing 45 percent in emerging markets.
On sustainability, approximately 72 percent of global consumers said they were willing to pay a premium for sustainable products, with most accepting a markup of 5 to 10 percent. The report devoted a dedicated chapter to artificial intelligence, noting that 58 to 60 percent of global fashion retailers have integrated AI into operations and marketing.
The report concluded that Shanghai Fashion Week is at a critical transition from scale expansion to capability upgrading, and should deepen integration of local culture, contemporary design and haute couture to strengthen its role as a commercial hub connecting design with consumption.
Original link: https://en.imsilkroad.com/p/350009.html
** This press release is distributed by PR Newswire through automated distribution system, for which the client assumes full responsibility. **

Xinhua Silk Road: Shanghai Fashion Week ranks 4th in latest vitality index report
HANGZHOU, China, April 3, 2026 /PRNewswire/ -- A research team led by Zhen-Xing Endowed Professor Jian Yang at the School of Life Sciences, Westlake University, together with collaborators, published their latest findings in Nature on April 1. The study innovatively developed a pangenome-informed genome assembly (PIGA) method. By combining a cost-effective hybrid sequencing strategy of long and short reads, the team successfully constructed a pangenome for over a thousand individuals. This achievement breaks through the limitations of previous small-sample pangenomes and provides a critical foundational infrastructure for medical and population genetics research.
Since the completion of the Human Genome Project, single linear reference genomes (such as GRCh38) have served as the foundation for biomedical research. However, the genetic backgrounds of human individuals vary significantly, and a single reference genome cannot capture the full extent of genetic diversity across populations. This leads to complex forms of genetic variations, such as structural variants (SVs) and tandem repeats (TRs), being overlooked in traditional analyses. To address this challenge, researchers proposed the concept of a pangenome—a collection of genome sequences representing the genetic diversity of a population.
While advancements in long-read sequencing have enabled the assembly of high-quality diploid genomes, the high costs of sequencing have limited the sample sizes of previous pangenomes to only a few dozen individuals. Such small sample sizes are insufficient to accurately estimate the frequency of genetic variants in populations or to resolve low-frequency variants and high-complexity regions. Therefore, developing a cost-effective pangenome construction strategy for large-scale populations has become an urgent requirement for resolving the functional impact of complex variants and enhancing clinical diagnostics.
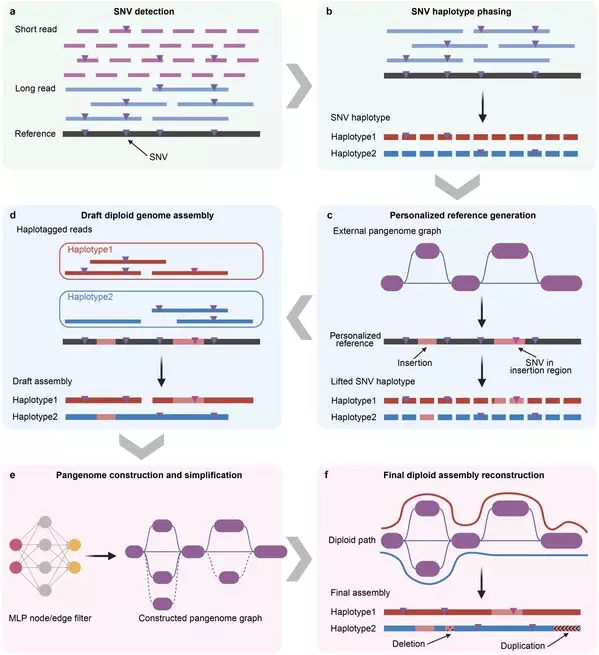
Yang's team has long been dedicated to methodological research in statistical genetics, genomics, and the big data analysis of human complex traits. By developing efficient computational methods, the team has consistently tackled core challenges in processing large-scale genomic data. Analysis tools developed by the team, such as GCTA-GREML, SMR, and gsMap, have been widely adopted globally. To address the challenge in constructing large-scale pangenomes, the research team developed the pangenome-informed genome assembly (PIGA) workflow (Fig. 1). Unlike de novo assembly approaches, which rely on sequencing data from individual samples, PIGA adopts a pangenome-guided framework to integrate sequence information across the entire cohort. It fully leverages a cost-effective hybrid sequencing strategy based on modest-coverage Illumina short-read and PacBio long-read whole-genome sequencing (WGS) data. This approach substantially reduces sequencing costs while enabling the assembly of genomes from modest-coverage data, thereby providing a practical new technical pathway for future population-scale hybrid sequencing studies.
Applying this method, the research team constructed the world's largest human pangenome to date, comprising 1,116 diploid genomes with a mean quality value (QV) of 46. The pangenome identified 405.3 million base pairs (Mb) of non-reference sequences absent from current references (GRCh38 and CHM13). Notably, the team annotated 26.2 Mb of these sequences as functional genic and predicted regulatory elements, greatly expanding our understanding of the non-reference sequences in the human genome.
Leveraging the large-scale assembly dataset, the researchers compiled a comprehensive catalog of genetic variation. In addition to 35.4 million small variants, the catalog captured a wide range of complex variants, including 110,530 SVs, 485,575 TRs, and 0.86 million nested variants embedded within non-reference sequences.
Using this catalog, the team characterized medically relevant variations at multiple scales (Fig. 2), including gene-altering SVs, pathogenic TR expansions, gene cluster variations, and HLA gene haplotypes. These findings indicate that the 1KCP variant catalog provides an important reference for the clinical screening of pathogenic mutations.
By integrating gene expression data, the team conducted pan-variant expression quantitative trait loci (eQTL) mapping. They identified 3,256 eQTLs involving complex variants (SVs, TRs, and nested variants), elucidating the regulatory complexity of these diverse variant types.
Together, this study significantly advances our understanding of complex genetic variants and their functional implications, establishing a new paradigm for human health research and pangenome studies in other species.
Ph.D. student Yifei Wang and Research Assistant Professor Zhongqu Duan are the co-first authors of the study. Professor Jian Yang is the last author. This work was supported by the National Natural Science Foundation of China, the National Key R&D Program, the Zhejiang "Pioneer & Leading Goose" Program, and the New Cornerstone Science Foundation. Computational resources were provided by the High-Performance Computing Center at Westlake University.
Professor Jian Yang's research group is dedicated to developing statistical genetics and bioinformatics methods. By deeply analyzing genomic and multi-omic data from large-scale population cohorts, they aim to uncover the genetic architecture and molecular mechanisms underlying complex diseases, translating these discoveries into novel strategies for disease diagnosis, drug target discovery, and precision medicine.
Related links:
Paper link: https://www.nature.com/articles/s41586-026-10315-y
Jian Yang lab website: https://yanglab.westlake.edu.cn/
Media contact:
Chi Zhang
media@westlake.edu.cn
+86-15659837873
** This press release is distributed by PR Newswire through automated distribution system, for which the client assumes full responsibility. **
Assembling Over 1,000 Human Genomes Affordably: New Method Powers Medicine's Future








